Maladie Hirschsprung (HSCR)
ou mégacôlon congénital
La maladie de Hirschsprung est une maladie congénitale rare qui touche 1 naissance sur 5000. Les nouveau-nés et les jeunes enfants sont les principaux touchés. Les symptômes et la sévérité peuvent varier d’un malade à l’autre.
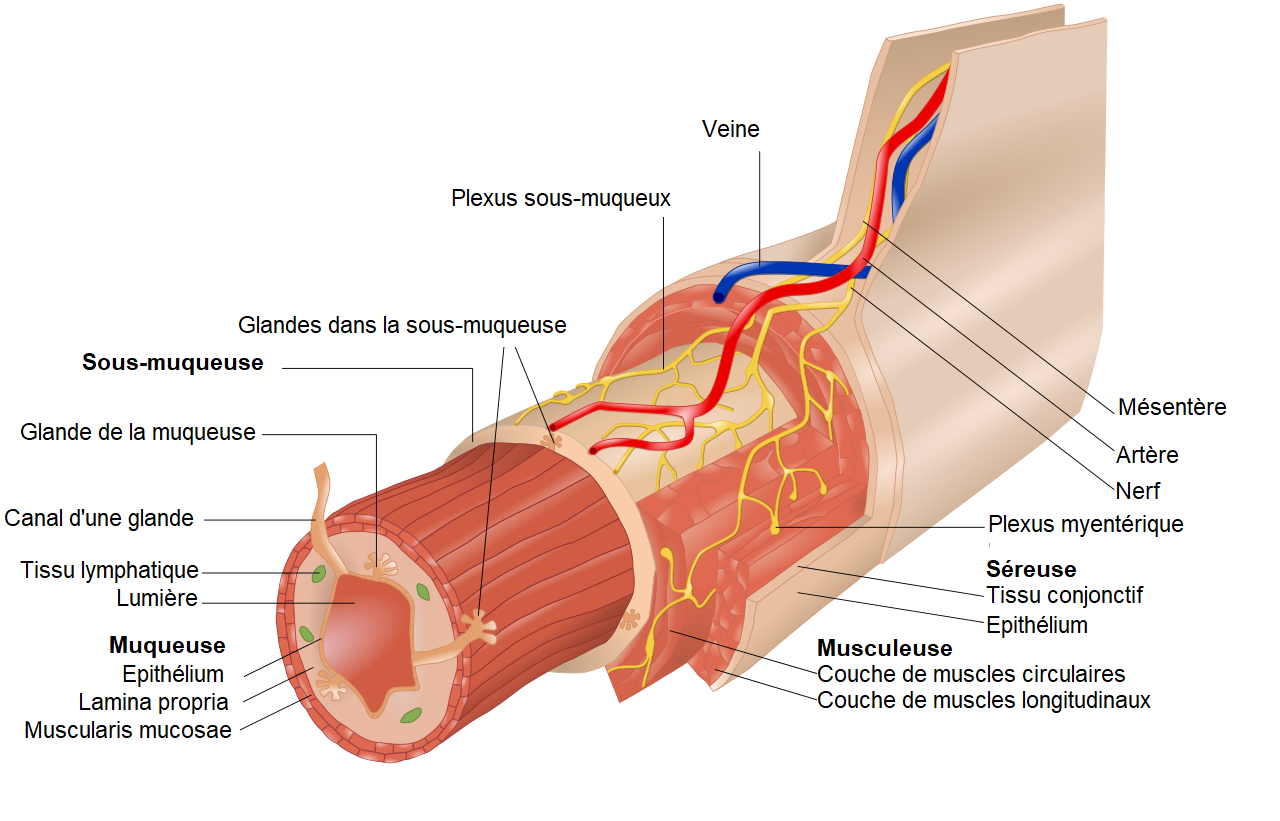
Dans cette maladie, le côlon (gros intestin) est atteint, il est dépourvu de ganglions nerveux dans sa paroi (aganglionose). Ces cellules ganglionnaires (cellules de Cajal qui sont les cellules pacemaker) constituent le système nerveux entérique (système nerveux de l’intestin) qui se trouve dans deux plexus (réseau de fibres nerveuses) :
- Le plexus sous-muqueux de Meissner qui contrôle les sécrétions digestives
- Le plexus myentérique d’Auerbach qui contrôle la motricité (péristaltisme) et donc le transit
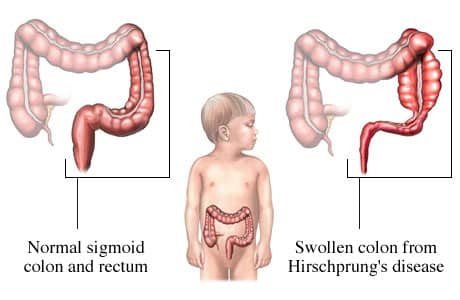
Atteinte sur le côlon
Paroi digestive
La maladie apparaît dès la naissance dans la plupart des cas, avec une absence de méconium (premières selles des nouveau-nés dans les 24h). Lorsqu’elle apparaît plus tard chez les enfants, un retard de croissance, une constipation sévère, une distension abdominale et une malnutrition sont au premier plan. Les nouveau-nés et enfants sont sujet à une infection intestinale grave appelée l’entérocolite qui nécessite une urgence vitale.
La gravité de la maladie dépend de la longueur du segment touché. En effet, Il existe 3 formes d’atteintes :
- La forme court segment ou recto sigmoïdienne (80%), la plus courante. Elle touche le segment terminal du côlon : du rectum jusqu’au sigmoïde. Les hommes sont plus touchés que les femmes.
- La forme long segment (15%), elle s’étend jusqu’en-dessus du côlon sigmoïde. Les filles sont davantage touchées par cette forme que les hommes.
- La forme totale (5%), elle touche l’ensemble du côlon et l’intestin grêle qui est également atteint. Il s’agit de la forme la plus grave.
Plus rarement, la maladie peut se révéler à l’âge adulte dont l’atteinte la plus fréquente est la forme « court segment ».
- Absence de méconium (premières selles) chez les nouveau-nés
- Distension abdominale et ballonnements
- Douleurs abdominales
- Crampes
- Occlusions intestinales
- Nausées, vomissements
- Constipation sévère ou gaz bloqués
- Diarrhée
- Absence de besoin d’aller à la selle
- Dénutrition
- Fièvre
- Congénital
- Facteurs génétiques: gène « RET »
- Mutations de gènes (multigénique)
- Biopsies chirurgicales superficielles de la paroi du rectum ou côlon pour déceler l’aganglionose
- Biopsies plus profondes pour confirmer le diagnostic si un doute subsiste
- Lavement au baryum (produit de contraste) : examen radiologique qui aide au diagnostic sur lequel l’intestin apparaît dilaté
- La manométrie anorectale : examen qui analyse avec une sonde et un ballonnet la pression des sphincters du rectum et mesure la sensibilité du rectum. Elle montre le relâchement incomplet du sphincter interne du rectum, disparition du réflexe recto-anal inhibiteur (RRAI) ne permettant plus à la matière fécale et aux gaz d’être expulsés
- La chirurgie permet d’enlever le segment de l’intestin atteint (aganglionose) et de raccorder le reste à l’anus (anastomose)
- Lavement ou irrigation colique (Peristeen) avec du sérum physiologique pour évacuer les selles
- Bouton de caecostomie ou appendicostomie percutanée
- Nutrition artificielle parentérale
- Iléostomie ou colostomie (ouverture faite chirurgicalement sur l’iléon ou le côlon, pour permette aux selles d’être évacuées dans une poche)
- Injection de botox dans le sphincter anal, pour aider au relâchement et à la matière fécale de descendre
Fig. 1 : https://www.accelerated-ideas.com/dicasdesaude/a-doenca-de-hirschsprung.aspx Fig. 2 : https://socratic.org/questions/from-the-lumen-outward-what-are-the-layers-of-the-gastrointestinal-tract